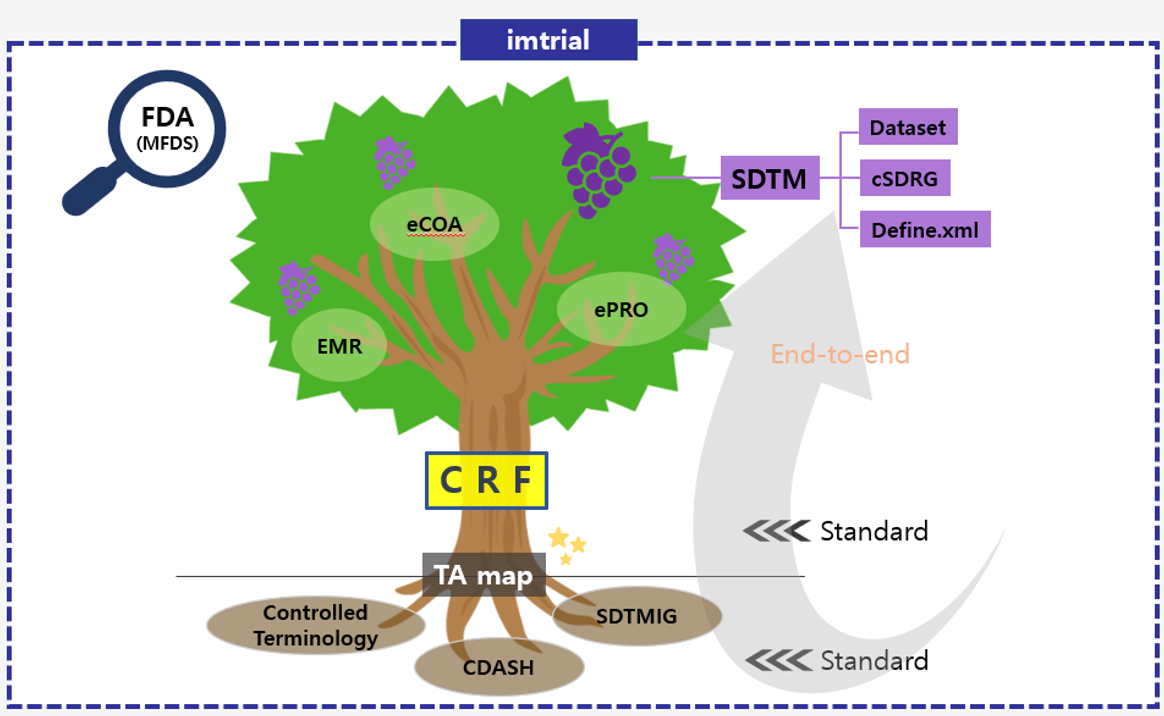
1. 배경
CDISC (Clinical Data Interchange Standards Consortium) 에서는 사람을 대상으로 하는 임상 시험자료 제출에 대한 표준을 정의하고 있으며 (SDTM), 이러한 표준을 활용할 수 있는 방법에 대한 가이드 (SDTMIG) 를 제시하고 있습니다. 글로벌 임상자료를 준비하는 제약사는 2가지 문서 즉, SDTM 과 SDTMIG 를 활용하여 FDA 에 제출하는 임상 데이터를 구성할 수 있게 됩니다.
CDISC 기준으로 SDTM 데이터를 구성했다고 하더라도, FDA 에 제출할 때는 FDA 에서 정한 규정에 맞게 제출되어야 하기 때문에 자료 제출 시 CDISC 는 물론, FDA 의 규정도 함께 검토해 보아야 합니다. CDISC 가 데이터의 수집 목적과 성격에 따른 표준 제출 방법에 대해서 자세히 제시하고 있다면, FDA 는 규제당국의 요구 및 검토 조건을 법령으로 제시하고 있습니다.
2. SDTM 과 관련된 FDA 규정
FDA 는 전자 문서를 제출하는 방법에 대해서 Section 745A(a) of the Federal Food, Drug, and Cosmetic Act 법에 제시하고 있으며, 이와 관련하여 아래와 같은 주요 가이드라인으로 나누어 제시하고 있습니다.
1) Guidance to industry Providing Regulatory Submissions in Electronic Format — Standardized Study Data
2) Guidance to Industry Providing Regulatory Submissions in Electronic Format: Certain Human Pharmaceutical Product Applications and Related Submissions Using the Electronic Common Technical Document Specifications
3) Study Data Technical Conformance Guide
https://www.fda.gov/industry/fda-data-standards-advisory-board/study-data-standards-resources
임상, 신약, 개량신약, 생물학적제제에 대한 허가를 받기 위한 전자 문서 검토 및 허가는 위 법령및 가이드 기준에 따라 제출되어야 하며, FDA 내 CDER (Center for Drug Evaluation and Research), CBER (Center for Biologics Evaluation and Research) 에서 검토 및 허가하고 있습니다.
3. Study Data Technical Conformance Guide 에 대한 이해
임상 및 비임상을 모두 포괄하고 있는 가이드는 계획, 수집 데이터, 제출에 관하여 순서대로 가이드를 제시하고 있으며, SEND, SDTM, ADaM 및 제출 자료에 대한 표준 형식 및 유의사항을 안내하고 있습니다.
1) Planning and Providing Standardized Study Data
Sponsor 는 SDSP (Study Data Standardization Plan) 를 제출해야 하며, 수집된 데이터 및 분석 데이터에 대해서 검토 부서에서 참고할 수 있는 리뷰어 가이드 cSDRG (Clinical Study Data Reviewer’s Guides), ADRG (Analysis Study Data Reviewer’s Guides) 를 제출하도록 규정하고 있습니다.
SDSP 는 전자 문서를 표준으로 제출하기 전에 어떤 방식으로 제출할 것인가에 대한 계획을 포함하는 문서로 반드시 제출해야 하는 문서입니다. SDSP 의 표지(Cover letter) 는 최신 버전을 표시해야 하며, 연구 중 계획이 추가되거나, 사용 프로그램이 확장될 때 마다 FDA 와 상의하여 문서를 업데이트하여 제출해야 합니다. SDSP 는 eCTD 내 1.13.9 General Investigational Plan 또는 1.20 General investigational plan for initial IND 에 위치해야 합니다. Sponsor 는 SDSP 의 내용에 대해서 Pre-IND 단계에서 논의할 수 있으며, Pre-NDA, pre-LBA 미팅 시 제공되어야 합니다. 만약 CBER 에 clinical data 를 제출 시 SDSP 의 appendix는 End-of-Phase (EOP2) 미팅 전 검토 부서에 제공되어야 합니다.
cSDRG 는 SDSP 의 계획된 내용과 제출된 데이터 간의 관계를 설명하는 리뷰어를 위한 가이드로, csdrg.pdf 이름의 PDF 형태로 제출되어야 합니다. eCTD m5 내 data-tabulation-data-definition 로 태그하여 위치합니다.
ADRG 는 제출된 데이터와 분석 데이터 간의 관계를 설명하는 리뷰어를 위한 가이드로 adrg.pdf 이름의 PDF 형태로 제출되어야 하며 eCTD m5 내 Aanlysis-tabulation-data-definition 로 태그하여 위치합니다. 이 때 유의할 점은 ADRG 를 제출해도 analysis dataset 의 define.xml 도 제출되어야 합니다. cSDRG 와 ADRG 는 반드시 제출되어야 하는 필수 문서는 아니지만 리뷰어가 데이터에 대한 이해를 돕기 때문에 제출을 추천하고 있습니다.
2) STUDY DATA
(1) 일반적인 사항
FDA 에 전자문서를 제출 시 XML, PDF, XPT 확장자를 가지는 파일 형식과 가이드에서 정한 이름으로 제출해야 합니다. XML 확장자를 가지는 파일은 압축 가능하지만 define.xml 은 압축해서는 안 되며, XPT 확장자를 가지는 파일은 압축되어서는 안 됩니다.
변수의 이름, 길이 등은 기본적으로 CDISC 에서 정한 표준을 따르는 것이 원칙이며, Dataset 이 5 GB 를 초과하는 경우 5 GB 아래로 쪼개어 제출해야 하고, split 폴더에 저장해야 합니다. 변수나 도메인에 특수문자는 ASCⅡ 만 포함 가능하며, 다만 LBSTRESC, LBTEST 에서 ASCⅡ코드 중 161번에서 191번에 해당하는 특수코드는 입력되어서는 안 된다는 사실을 유의해야 합니다.
FDA 규정은 CDISC 에서 제시한 SDTM 문서 외 추가로 필요한 분석 파일을 별도의 문서나 SDRG 에 솔루션을 제출하는 것을 적극 추천하고 있습니다. Define.xml, aCRF 에 대한 세부 유의사항도 포함하고 있습니다.
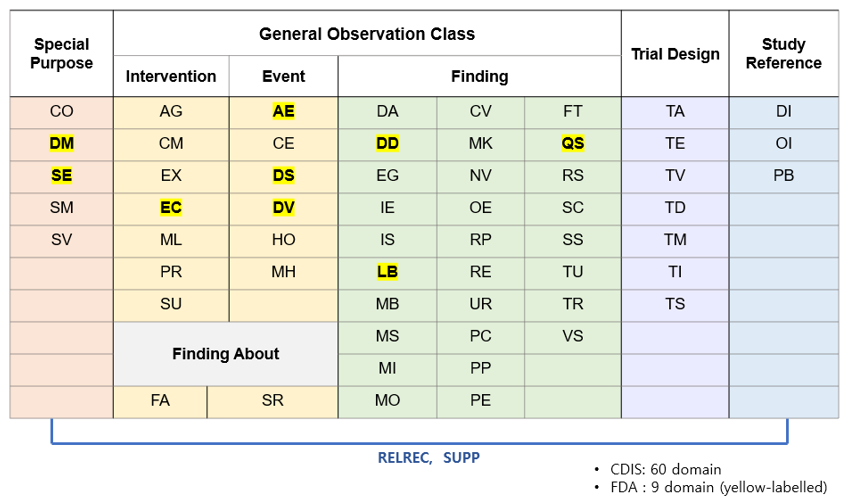
(2) 도메인 별 사항
FDA 규정에서는 CDISC 에서 제시한 표준 60개의 도메인 모델 중에서 자료 검토 시 필요한 9개 도메인에 대한 유의사항을 언급하고 있습니다. 예를 들어 DV (Protocol Deviations) 도메인의 경우, DV 도메인을 반드시 제출하도록 안내하고 있으며, CDISC 에서 required variable 로 정한 DVTERM 외에 permissible variables 인 DVSPID, DVDECOD, DVCAT, DVSCAT, DVSTDTC, EVENDTC, EPOCH 를 반드시 포함하는 것으로 규정하고 있으니 자료 제출 시 FDA규정을 참고하여 SDTM 자료를 마련해야 합니다. 이외 도메인 별 해당하는 세부사항은 해당하는 도메인이 있을 경우 FDA 규정을 리뷰해 보는 것을 추천 드립니다.
(3) TERMINOLOGY
표준으로 자료를 수집 시 중요한 부분 중의 하나는 용어집을 통한 일관된 용어를 사용하는 것입니다. SDTM 에서 사용하는 용도는 데이터의 수집 목적에 따라 여러 용어집을 활용하도록 규정하고 있으며, 아래와 같이 분류됩니다.
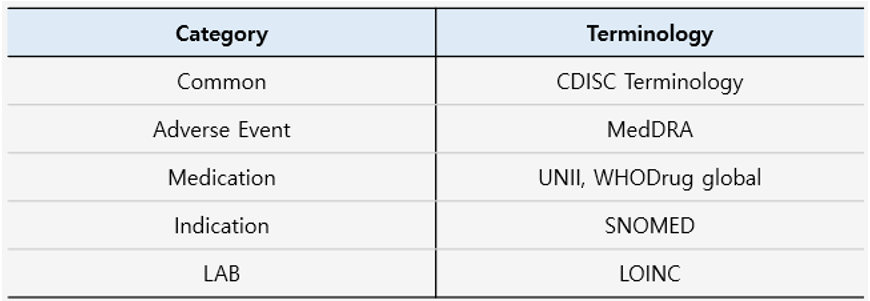
일반적으로 SDTM 자료를 수집할 때는 CDISC 용어집을 따르지만 이상반응을 수집할 때는 MedDRA (이상 반응에 관한 분류사전, 국제 의학 용어집) 를 사용합니다.
약물 처방에 관해 허가 받지 않은 임상약의 경우 성분을 식별할 수 있는 UNII (고유성분식별자) 혹은 의약품 정보가 있는 경우 WHODrug global 를 사용합니다. Indication 이 있을 때는 SNOMED (EHRs-전자건강기록지-에 사용할 수 있는 다국어 임상 의료 용어 체계) 를 사용하며, 실험실 테스트의 경우 LOINC (진단검사 표준용어) 에 맞춰 SDTM 자료를 제출하고, TS Domain 에 활용한 용어집 이름과 버전을 기록해야 합니다. 일반적으로 가장 최신화된 용어집 버전을 사용해야 합니다.
3) SUBMISSION
파일의 형식, 크기, 제출 방법 등을 CDISC 에서 제시한 SDTMIG 및 FDA 규정에 모두 맞게 최종적으로 dataset 이 마련된 경우, 전자적 문서 형태로 FDA 에 eCTD 문서로 제출하면 됩니다. 제출 시에는 허가를 받기 위한 많은 문서가 있지만, 이번 가이드에서는 SDTM 에 관해 중점적으로 다루고 있으므로 전체적인 제출의 목록에 대해서는 다루지 않으며, eCTD 로 SDSP, cSDRG, ADRG 에 대한 부분 및 SDTM 자료 제출에 대한 내용만 언급하는 점을 유의하시기 바랍니다. 가이드와 관련된 전자 문서는 module5 폴더 및 SDTM 관련 문서는 하위폴더인 dataset> tabulation> SDTM 및 split 에 저장하여 제출하면 됩니다.
4. Conclusion
지금까지 SDTM 데이터를 FDA 로 제출할 때 고려되어야 하는 사항 및 전자적 문서 제출에 대한 규정을 전반적으로 확인하고 공유하였습니다. CDISC 에서 제시하는 일반적인 전자 데이터의 생성 방법에 따른 SDTM 자료는 또한 FDA 규정에도 적합해야 한다는 것을 알 수 있었습니다.
imtrial 은 CDISC 에서 제시하는 방법에 맞게 전자 데이터가 자동으로 변환되어 SDTM 문서로 생성될 수 있음은 물론, FDA 규정에서 살펴본 일반적인 사항, 도메인 별 상세한 유의사항에 대한 내용도 프로그램에 반영되어 있어 FDA 에 자료 제출 시 발생할 수 있는 미연의 실수를 사전에 차단할 수 있어 빠르고 효율적인 자료 생성이 가능합니다.
또한, 프로그램 내부에 FDA 에서 규정하는 Catalog 의 최신 버전과 Controlled Terminology 가 구축되어 있어 수집 단계에서부터 효율적인 프로세스로 자료 생성 및 관리가 용이하며, FDA 규정에 따른 전자적 문서인 cSDRG (PDF파일) 이 자동으로 생성되어 sponsor는 별도의 준비 없이 imtrial 을 사용하는 것만으로도 SDTM dataset 과 함께 규제당국이 원하는 cSDRG 가 마련됨은 물론 define.xml 자료도 생성되어 하나의 플랫폼으로 다양한 자료가 동시에 생성되는 것을 경험할 수 있습니다.
imtrial 플랫폼은 하나의 자료를 입력하는 것 만으로 단순히 SDTM 자료를 제공하는 것 뿐 아니라 FDA 규정에 맞는 다양한 문서에 고도화된 자료를 제공해 드리고 있습니다.